
Let's try GAMESS on the simplest system where everything began, the hydrogen atom.
If the energy is not 13.6 eV, then it is wrong.
The goal of this tutorial is to make a GAMESS input file for the hydrogen atom and then run it on your computer system. This tutorial uses GAMESS on Windows 64 bit. If you are using another operating system, the input file will be the same but the process to run the calculation maybe a little different.
1. Make input file
We will write the input file using notepad (I suggest to use notepad++).GAMESS input file contains several input groups. An input group starts with $[input group name] and ends with $end. (You should read GAMESS documentation to get a detail description of each input group.) Some input groups are required while others are optional. Here we only use three input group:
$contrl
This group specifies fundamental job options. Here I write the commands and their explanation.$contrl runtyp=energy scftyp=rohf mult=2 $end
Note: You should put a blank (space) in front of a control group command. GAMESS will only run them if you do so.
- runtyp=energy
RUNTYP specifies the type of computation.
ENERGY means this is a molecular (or atomic in our case) single point energy calculation. It is the default option. - scftyp=rohf
SCFTYP specifies the self-consistent field wavefunction.
ROHF is Restricted open shell Hartree-Fock. We use this because our system has duplet (2) multiplicity. - mult=2
This gives information about our spin multiplicity. We can calculate it using 2S+1 with S is the total spin of our system.
$basis
This group allows certain standard basis sets to be easily requested. In this calculation, we use a Gaussian basis set: Pople's "triple split" N-311G. This basis set available for NGAUSS=6. The command for it is as follows.$basis gbasis=N311 ngauss=6 $end
$data
This group describes the global molecular data. It contains several lines as follows.$data
My first hidrogen input
C1
H 1.0 0.0000000 0.0000000 0.0000000
$end
- The second line is TITLE: a description of your calculation (use only for printout).
- The third line is GROUP: the Schoenflies symbol of the symmetry group. We use C1 which means no symmetry.
- The fourth line is ATOM INPUT. We fill it with: [Atom symbol/name (use only for printout)] [Nurclear charge] [X Y Z (cartesian coordinate)]
Our input file is ready now. It should look as follows.
$contrl runtyp=energy scftyp=rohf mult=2 $end
$basis gbasis=N311 ngauss=6 $end
$data
My first hidrogen input
C1
H 1.0 0.0000000 0.0000000 0.0000000
$end
Now, save your file with .inp extension.
2. Run the calculation
After installing GAMESS on your computer system, you should already follow the tutorial on the Readme file to run a sample input file using rungms.bat script. In this tutorial, we will use double-click-run.bat script.
Step 1
Copy your input file to C:\Users\Public\gamess-64\inputs
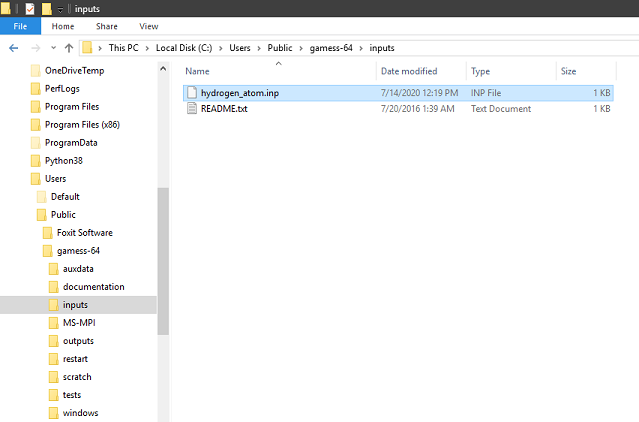
Step 2
Click file double-click-run.bat on C:\Users\Public\gamess-64 then the calculation will be automatically begun. Wait until message Finish with GAMESS job submission appears, then press any key to close the dialog box.
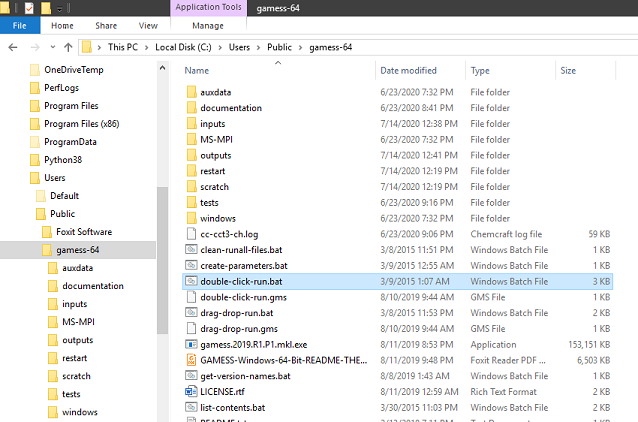
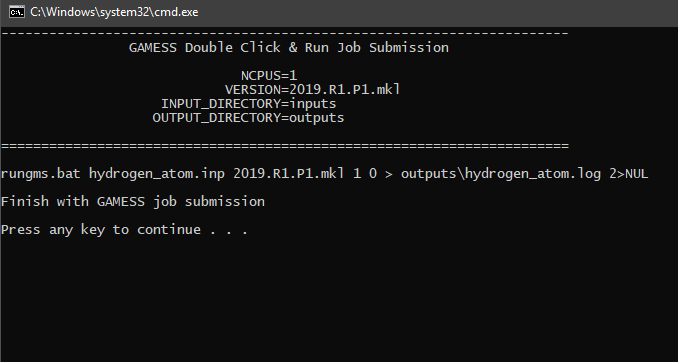
Step 3
You can find your output file at C:\Users\Public\gamess-64\outputs.
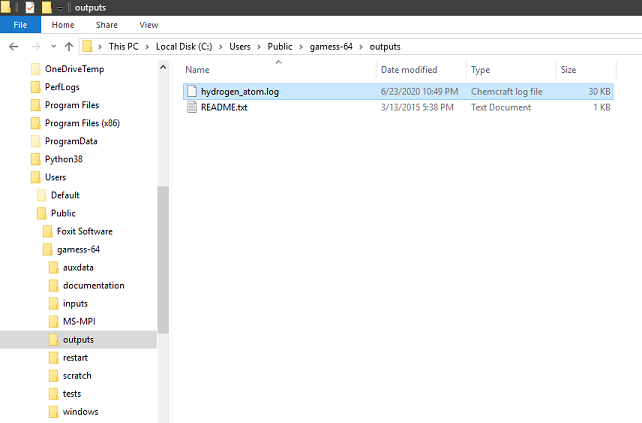
Alterlative
If you want to run the calculation from another folder (outside the installation folder), copy file double-click-run.bat, double-click-run.gms, rungms.gms and folder inputs, outputs to that folder.
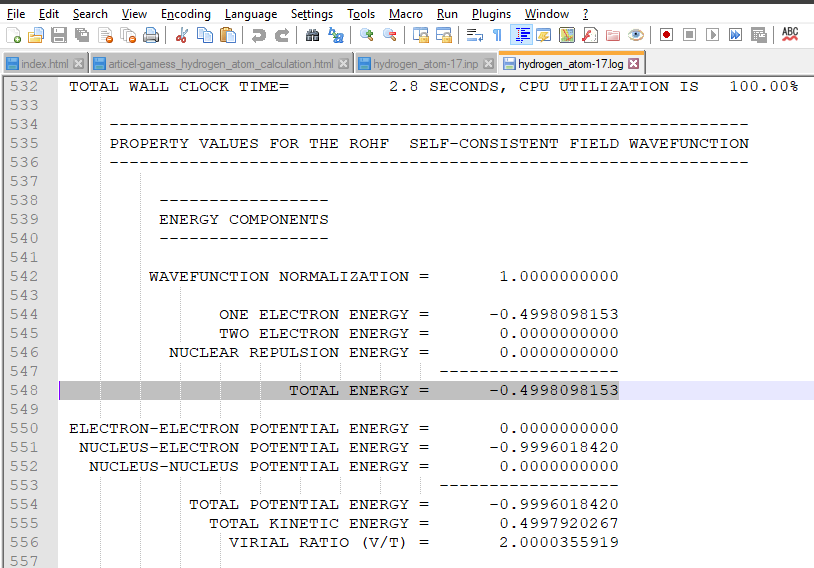
Now let see our output file. Open it use notepad++. Find line that contains total energy. It is the total energy of our system (in hartree units). Convert it into electron volt and if there is nothing wrong, it should be 13.6 eV.
That's all, see you around!!!